Stereochemistry and Atom Parity in SMILES
SMILES notation supports the reading and writing of tetrahedral stereochemical configuration. Usually the job of dealing with this notation falls to software. But sometimes it falls to you, the chemist or software developer. This article explains the SMILES stereochemical notation system in detail.
Atom Parity
SMILES expresses stereochemical configuration through atom parity. Atom parity is a boolean (true/false) value that designates a stereocenter as either having an identical or opposite configuration to a reference.
The most widely-used atom parity system is Cahn-Ingold-Prelog (CIP). Substituents on a tetrahedral stereocenter are ranked according to a multi-level system of "priority rules" (aka "sequence rules"). The stereocenter is then oriented to place the lowest-priority substituent behind the stereocenter. If a curve passing through the remaining substituents winds clockwise, the center is assigned the parity label (R). Otherwise the label (S) is applied.
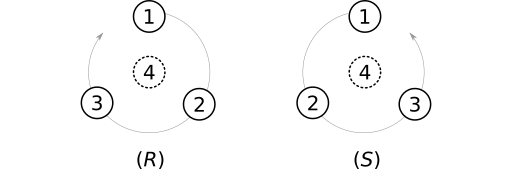
SMILES adopts a similar system. Most noticeably, SMILES uses the labels @ (counterclockwise) and @@ (clockwise), which were designed as mnemonics. The at symbol (@) winds counterclockwise from the center out. The label @@ brings to mind counter-counterclockwise, or clockwise, winding. Similar mnemonic origins apply to the CIP R (rectus, or "right") and S (sinister, or "left") labels.
Fortunately, the priority rules in SMILES are simpler than those for CIP. But before getting to that, one more simplification will be helpful.
Stereocenter Syntax
An atom using a parity label must be enclosed with brackets ([ and ]). Semantically, this notation also requires explicit encoding of virtual hydrogen counts greater than zero.
In practice, the virtual hydrogen count can't exceed one. A tetrahedral stereocenter requires a total of four unique substituents. One can be virtual hydrogen, leaving three that must be unique. But if two virtual hydrogens are present, they are by definition equivalent and so may not be attached to a stereocenter.
Additional constraints follow from the SMILES grammar, which provides a concise set of "production rules" for reading and writing SMILES strings. Consider the production rule for a bracket atom:
<bracket_atom> ::= "[" <isotope>? <symbol> <parity>? <hcount>? <charge> <map>? "]"
The two most important nonterminals (items in brackets) here are <parity> and <hcount>. <parity> is one of the atom parity labels @ or @@. <hcount> is the character H followed by an optional digit (0-9). But as explained previously, the hydrogen count on a stereocenter must be zero or one.
Combining the bracket atom syntax rule with the semantic rules around hydrogen count allows us to conclude that a tetrahedral SMILES stereocenter will occur in one of the following two forms:
@or@@. Four atomic neighbors will be present. A less-common expression of the same notation would be@H0or@@H0.@Hor@@H. Three atomic neighbors and a virtual hydrogen will be present. A less-common expression of the same notation would be@H1or@@H1.
Referring back to these two forms will simplify the process of manually encoding and decoding stereo SMILES strings.
Priority Rules
Having determined the forms that stereochemical notation will take, let's move on to the SMILES priority rules. In CIP, these rules are multifaceted. In SMILES, however, priority is based on just one rule: the order of connection.
Think of a SMILES string as a left-to-right instruction sequence for building a molecule. Atoms are added and connected through bonds in their order of appearance within the string. Parentheses are always traversed greedily. Virtual hydrogens are connected immediately on encountering them. Similarly, ring closures are connected in their order of appearance.
From this principle it follows that connection order to a stereocenter follows the relative order in which bonds appear within a SMILES string. Given two bonds to a stereocenter, the one to the left will always lead to higher substituent priority than the one to the right.
SMILES allows certain bonds to be elided. For example, the SMILES string CC contains one elided bond between two carbon atoms. Elided bonds are treated the same as any other bond. The further to the left a bond's presence is implied, the higher the priority of its associated substituent. This rule applies to atoms, ring cuts, and virtual hydrogens equally.
Consider 1-aminoethanol. Unstable though it may be, we can nevertheless encode its enantiomeric forms using SMILES. One one of them could be written as:
[C@H](O)(N)C
The order of substituent priority about the stereocenter is: H; O; N; C. Hydrogen (as a virtual hydrogen) is attached first, followed by oxygen, nitrogen, and finally carbon.
Another formulation of the same enatiomer can be written as:
O[C@@H](N)C
In this case, the order of substituent priority follows the sequence: O; H; N; C. Notice that atom parity has been flipped to preserve the sense of chirality. Rules for making such conversions will be given shortly.
To recap: The priority of a substituent is the relative order in which its bond to the stereocenter appears in the SMILES string. There are no exceptions.
Reading and Writing Parity
After substituent priorities have been determined, atom parity can be assigned using these steps:
- Identify the substituent with highest priority. I'll call this the prime atom (Weininger uses the sometimes hard-top-parse term "from atom").
- Place the prime atom between yourself and the stereocenter.
- Trace a curve through the remaining three substituents, starting with the one having the highest priority.
- If the curve winds clockwise, apply the parity label
@@. Otherwise, apply the label@.
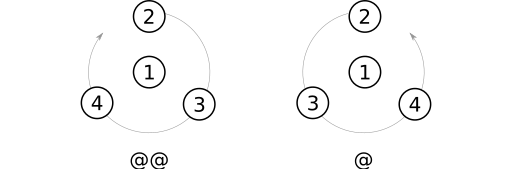
@@ parity label, or the @ label otherwise.
These rules can be used to either read or write the configuration of a stereocenter. When reading, we transform a label (@ or @@) into a stereochemical configuration. When writing, we transform a configuration into a label.
Imagine reading the single-enantiomer SMILES representation for 1-aminoethanol, [C@H](O)(N)C. Substituent priority follows the order: H; O; N; C. The "from atom" is therefore H. Sighting down H toward the stereocenter, we wind the remaining substituents counterclockwise in agreement with the label (@). We can generate a 2D stereo view (wedge/hash style) by 90˚ rotation of the perspective to the left.

[C@H](O)(N)C, priority follows the order: H; O; N; C. Orienting the prime atom (H) over the stereocenter, wind the remaining substituents in counterclockwise (@) order (left). Rotating 90 ° left reveals a directly displayable 2D representation (right).
The reverse process can be used to write a SMILES string from a 2D or 3D stereo view. Consider (R)-bromochlorofluoromethane. Begin with any achiral SMILES representation — for example [CH](Br)(Cl)F. The priority order is: H; Br; Cl; F. Orient the 2D model so that H (the prime atom) is in front of the stereocenter. Next, note the counterclockwise winding of the remaining substituents in the order Br, Cl, F. Therefore, the parity label will be @@ and the complete SMILES is therefore [C@@H](Br)(Cl)F.
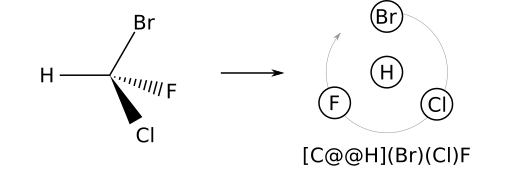
@@ should be used.
Transformations
It's sometimes necessary to compare stereo SMILES for equivalence. One way to do so is through an intermediate 2D or 3D view. For example, the SMILES Depicter can generate 2D stereo views. As stereochemical support continues to be added, another 2D viewing option would be the ChemWriter SMILES page. A 3D virtual tool or a physical model set could also be used.
But some situations call for a more direct approach. For example, complex SMILES may generate coordinates that will be difficult to compare on a screen due to differences in overall orientation. Alternatively, your goal may be to build a software tool to compare or transform SMILES sterecenters. Maybe you just want a more algorithmic way to compare stereo SMILES strings. In these cases, what's needed is a set of rules for interconversion.
Five primitive operations will transform a stereo SMILES string in useful ways:
- Virtualize. Moves an atomic hydrogen on the immediate right of the stereocenter inside the brackets.
- Reify. Moves a virtual hydrogen into the immediate right position.
- Swap. Exchanges any two substituents to the right of the stereocenter, and flips the parity label.
- Slide Left. Moves a substituent on the immediate left of the stereocenter to the immediate right position. Disabled if the stereocenter carries a virtual hydrogen.
- Slide Right. Moves a substituent on the immediate right of the stereocenter to the immediate left position. Disabled if the stereocenter carries a virtual hydrogen.
Using these operations, any stereo SMILES can be converted into a more convenient form.
For the purpose of reasoning about atomic configurations encoded within stereo SMILES strings, I use a form I call stereocentric. In stereocentric form, the stereocenter of interest appears at the beginning of the SMILES string. Hydrogen, if present, is virtualized. This translates to a top-down visualization of the stereocenter, with the prime atom eclipsing the stereocenter and the remaining substituents arranged either clockwise or counterclockwise. Creating a 2D depiction from a stereocentric SMILES is simple.
Consider (S)-alanine, represented as the SMILES O=C(O)[C@@H](N)C. It can be recast into stereocentric form with the following operations:
- reify:
O=C(O)[C@@]([H])(N)C - slide right:
[C@@](C(=O)O)([H])(N)C - swap:
[C@]([H])(C(=O)O)(N)C - virtualize:
[C@H](C(=O)O)(N)C
This form can be drawn in 2D by winding the substituents counterclockwise in the order: C(=O)O; N; C.

Beware of Ring Closures
No special rules are required for ring closures. Nevertheless, special care is called for when working with them. In particular, it's important to remember that priority is determined by the order in which bonds appears in a SMILES string, not the order in which atoms appear.
Consider (S)‑2‑fluorooxirane, which can be represented by the stereocentric SMILES F[C@H]1CO1. Recall that a ring open adds a bond to the stereocenter immediately when encountered. For this reason, the substituent priority order is: F; H; O; C. A form more amenable to manual depiction can be obtained with the operations: reify; slide right; swap; virtualize. The result is [C@@H](F)1CO1.
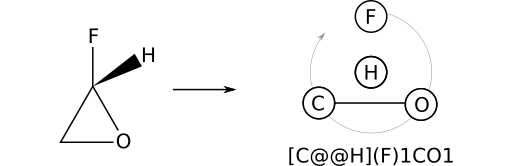
To re-iterate, substituent priority does not track the order in which a substituent appears in a SMILES string, but rather the order in which the substituent's bond to a stereocenter was added. In the case of (S)‑2‑fluorooxirane (F[C@@H]1CO1), the ring bond to oxygen is added before the bond to carbon (1 precedes C). This leads to the priority order F; H; O; C.
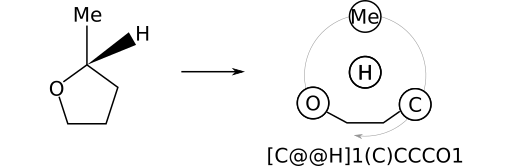
As another example, consider (R)‑2‑methyltetrahydrofuran, whose stereocentric SMILES can be written as [C@H](C)1CCCO1. The substituent priority order is: CH3; O; CH2. Again, this order follows from the order in which bonds containing the stereocenter's substituents appear in the SMILES string.
@@ parity label (right).
Other Sources
Although the OpenSMILES page documents SMILES stereochemistry to a degree, I found that the treatment left too many questions unanswered. The description of stereochemistry by Weininger is worth reading. Recently, Noel O'Boyle offered some thoughts on how Open Babel deals with stereochemistry. Somewhat unexpectedly, the BIOVIA Chemical Representation Guide contains a discussion on atom parity that I found very useful in the context of SMILES.
Conclusion
SMILES represents stereochemistry using a system comprised of parity labels and substituent priorities. Virtual hydrogens, ring cuts, and atoms can all be handled in the same uniform way. Rules for interconverting SMILES — while retaining stereochemical configuration — simplify the task of comparing atom configurations and producing 2D/3D representations.