Chemception: Deep Learning from 2D Chemical Structure Images
Recent advances in machine learning techniques have yielded systems that meet or even exceed human pattern-recognition capability. These powerful techniques are now starting to be used for chemical structure-property prediction. This article highlights a new approach that, in a break with past systems, works in a way that will be immediately (perhaps uncannily) recognizable to many research chemists.
Chemception

Chemception is a deep convolutional neural network (CNN) that accepts 80x80-pixel 2D chemical structure images as input. It can be configured to produce binary classifications ("active" vs "inactive" predictions) or regressions (numerical predictions) as output. The original Chemception paper describes training and use on datasets drawn from three areas: a panel of vitro toxicity assays (Tox21); an in vitro anti-HIV assay; and hydration free energy (FreeSolv).
Chemception's accuracy compared favorably with, and sometimes exceeded, existing machine learning and computational methods. However, those other methods required extensive involvement from trained subject-matter experts. Chemception, on the other hand, required only the transformation of chemical line notations (SMILES strings) into appropriately-encoded grayscale images.
Chemception demonstrates that a deep neural network trained on 2D structure images can make predictions that rival or even exceed those from systems driven by human-driven feature identification.
Image Encoding
Grayscale 80x80-pixel images were created directly from SMILES strings using a pipeline of open source tools. Atoms were encoded as dots with a shade of gray proportional to atomic number. Bonds were were encoded as lines between atoms using a shade of grey proportional to the number 2. The remainder of the image was colored black (a value of zero).
The authors don't discuss whether or not bond order was encoded in any way. Indeed, the description seems to state that bond order was not considered at all. If true, this lack of chemical information makes the relatively high accuracy of Chemception's predictions all the more impressive.
Images were barely processed further before entering the Chemception pipeline. Noting the sparsity of information density (~90% of pixels were set to zero in any given image), various helpful transformations used in other image classification studies were not performed. Images were randomly rotated 180 degrees, but were otherwise used as-is. A subsequent study (see below) encoded additional chemical information using a four-channel model.
Machine Learning vs. Machine Intelligence
The creators of Chemception identify two fundamentally different modes for machine-assisted chemistry research:
- Machine Learning Approach. Molecular features are selected and encoded through human-developed algorithms. These features are then used to train an artificial structure-property prediction system.
- Machine Intelligence Approach. Molecular features are selected and encoded automatically during the training of a structure-property prediction system.
The development of predictive models in chemistry has been dominated by the first approach. Chemists apply their deep domain knowledge to define those molecular features deemed important for predicting a molecular behavior. These features, usually in the form of "molecular descriptors," are then used to train a model. The development of molecular descriptors (of which extended connectivity/circular fingerprints are an example) has been ongoing area of research since the 1940s with well over 3,000 having been described to date.
By accepting raw input in the form of 2D structure images, Chemception eliminates the need for the human subject matter expert during training. Considering the astonishing rate at which artificial pattern recognition has caught up to and exceeded human capability in areas like image recognition, the economic and scientific implications of the Machine Intelligence Approach embodied by Chemception are provocative.
Deep Neural Networks
Chemception is an example of an artificial neural network (ANN). Neural networks come in a variety of configurations, but all can be modeled as directed graphs in which nodes map to functions, inbound edges map to arguments, and outbound edges map to return values. A neural network is typically arranged into layers of nodes disconnected from each other but connected to nodes in adjacent layers. At a minimum, an ANN contains two layers: an "input layer" that directly receives test or training data; and an "output layer" producing a classification or regression output. Layers between input and output are known as "hidden layers." By one widely-used definition, an ANN containing two or more hidden layer is considered "deep." Machine learning that uses a deep neural network is called "deep learning."
Chemical structure-property machine learning studies often use a kind of an ANN configuration known as a multilayer perceptron (MLP, aka "vanilla" neural network). An MLP is characterized by at least one hidden layer, the nodes of which connect to all of the nodes in adjacent layers, but which do not connect to each other. Excellent introductions to the structure and function of MLPs are available in the video series embedded above and in the free book Neural Networks and Deep Learning. The image below, excerpted from the book, illustrates the organization of layers in an MLP.

When applied to chemistry, MLPs typically accept binary fingerprints as input.
Convolutional Neural Networks
Chemception uses a kind of augmented MLP known as a convolutional neural network (CNN). A CNN employs one or more convolutional layers immediately following the input layer. In contrast to the nodes in the hidden layer of an MLP, nodes in a convolutional layer only connect to a subset of the nodes in the preceding layer. A convolutional layer detects patterns in the input layer through the stepwise application of one or more "filters" during a sweeping "convolve" operation.
CNNs have proven extremely adept at image recognition and for this reason are currently the subject of intense interest. One of the most successful designs (Inception) was integrated into Chemception.
Estimating Accuracy
Two of the predictions made by Chemception, HIV activity and toxicity, were binary classifications (molecules were marked as either "active" or "inactive"). The accuracy of such classifications can be estimated by measuring the area under the curve (AUC) of the Receiver Operator Characteristic (ROC) graph. This graph plots the false positive rate (a number in the range 0.0-1.0) on the x-axis and the true positive rate (a number in the range 0.0-1.0) on the y-axis for varying classification thresholds. AUC ranges from 0.0 (model only predicts false positives) to 1.0 (model only predicts true positives). The video below explains AUC in detail.
For example, the Chemception paper reports AUC for several configurations of the neural network tested against the Tox21 dataset (below). This study revealed one network configuration (Chemception_T3_F16) that worked the best. Additionally, the comparable AUCs for Train, Validation, and Test modes suggests that little overfitting occurred.

Performance Benchmarks
Using ROC AUC, Chemception's performance was compared to that of a previously-reported multi-layer perceptron deep neural network (MoleculeNet). In that study, extended connectivity fingerprints were inputs. The prediction accuracies for both systems were comparable, as shown in the graph below.

Even better results were obtained against the same MoleculeNet network using the HIV dataset (below).
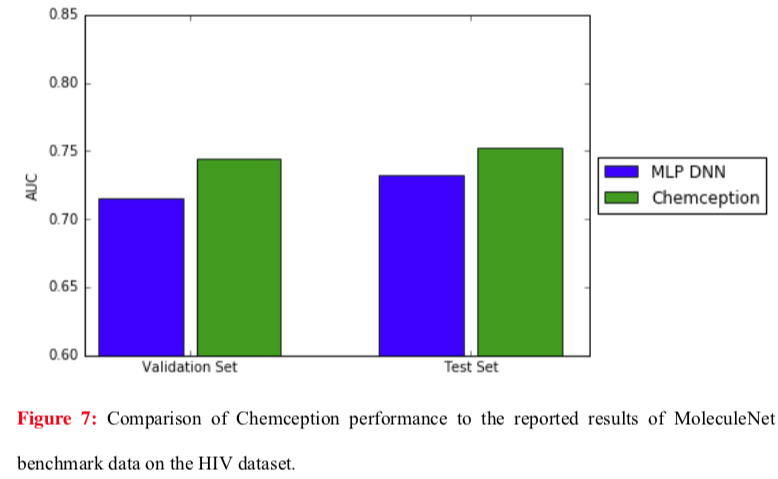
The Chemception paper notes two points that may account for the differences between the Tox21 and HIV studies. First, the Tox21 comparison represents a worst case for Chemception because MoleculeNet was operating in multi-task mode, giving it a theoretical advantage. (Recall that the Tox21 dataset is comprised of a panel of twelve assays.) The ideal comparison would have pitted MoleculeNet against Chemception in single-task mode, but single-task data were not reported in the MoleculeNet study. Second, the HIV dataset is larger than the Tox21 dataset. Larger datasets are expected to help Chemception given that it must synthesize all of its own chemical feature identifications.
Chemception's performance in regression against published techniques was also evaluated for the FreeSolv hydration free energy dataset. Here, two previous benchmarks were compared using root mean square (RMS) analysis: MoleculeNet and FEP (molecular dynamics simulation). The results show Chemception outperforming MoleculeNet, and approaching the performance of the molecular dynamics simulation.

AugChemception
The most surprising result from the Chemception study was how little chemical information was needed to produce competitive predictions. This raises the question of whether more information-rich 2D structure images might perform better. To address this question, the Chemception team conducted a follow-up study in which a new system, AugChemception was developed and tested.
Using up to four color channels, 2D structure images were augmented with four chemical information properties:
- bond order
- partial charge
- atom hybridization
- valence
These properties were encoded into four images channels in analogy with the standard CMYK model. Ten schema were developed to aid in understanding the minimum chemical information requirements for effective model training:

For example, "Std" represents the encoding used in the original Chemception paper: atomic numbers were encoded as shaded dots using a normalized grayscale. Bonds were assigned the relative grayscale 2 (a convenient choice given the lack of helium-containing structures). "RedA" images contained the least structure information because only atom dots were rendered, and each with the same shade of gray. "RedB" extends RedA with bond lines of uniform grayscale value. "EngA" encodes structural properties over four color channels. The last three schemes represent controls designed to detect artifacts and overtraining. Although the AugChemception paper doesn't give good examples of what these images actually look like, a subsequent study, described below, does.
The "engineered" schema EngA, EngB, EngC, and EngD yielded clear increases in accuracy for all datasets tested. For example, against the Tox21 dataset, the engineered AugChemception images performed about 4% better in terms of ROC AUC compared to the grayscale "Std" Chemception scheme. Similar results were obtained against the HIV dataset.

An apparent split in accuracy favoring EngA/EngD over EngB/EngC schemes against the hydration free energy dataset was noted. Although all four schemes outperformed the original Std Chemception grayscale scheme, EngA/EngD performed the best of all. The authors note:
Upon further examination, both EngA and EngD had partial charge and hybridization information encoded, but EngB and EngC do not.
Indeed, it can be seen from Table 2 that EngA/EngD encode both partial charge and hybridization. EngB/EngC only encode one property or the other. The combination of visual cues for both partial charge and hybridization appears to yield a significant increase in accuracy. Unfortunately, no scheme encoding just partial charge and hybridization was included in the AugChemception study.
Perhaps the most interesting finding of the AugChemception study was the one involving RedA. This schema only encodes atom positions with a uniform grayscale value of 1. In other words, these images consist of nothing more than uniformly shaded points representing the relative positions of atoms laid out on a 2D coordinate system.
Referring back to Figure 3, which records accuracies against Tox21, RedA produces about 72% AUC. While this is significantly below the original Chemception Std schema, it is well above the 0.5 AUC for random noise. Although RedA performed much more poorly against the other two datasets, its performance in Tox21 demonstrates that images carrying no chemical information other than 2D atom layout can produce models much more accurate than noise and within the range of more chemically-rich schemas.
As the authors note:
These results suggest that explicit knowledge of bonds was apparently not the most important requirement for determining molecular toxicity. Given how fundamentally important the concept of chemical bonds is in chemistry, at first glance this observation is paradoxical. However, one has to recall that a bond is an artificial construct introduced to denote the linkages between various atoms, and one of its key role in chemistry research is to make it easier for chemists to formulate more sophisticated concepts starting with the notion of a bond.
The best AugChemception schema was then compared with reported model building systems. The results show AugChemception to be competitive across the board.

Whither Fingerprints?
Chemception offers a compelling demonstration of the power of CNNs to extract chemically-meaningful features with very little human guidance. However, it's not alone. Within the last two years, at least two other deep learning systems have been reported that forgo molecular fingerprints for more direct graph representations:
- ConvGraph. Implements convolutional layers designed for use on graph representations directly.
- Continuous Representations from SMILES. Maps an arbitrary raw SMILE string to a coordinate in continuous "latent space." After translation by some distance, the new point can be decoded to a new molecular representation.
Given the novelty of these fingerprint-free approaches and large space for optimization, it seems likely that systems based on this new paradigm will continue to advance and may one day begin to consistently outperform fingerprint-based methods.
Hands-On Practice
A recent tutorial by Esben Bjerrum reported a detailed, step-by-step procedure for creating and training an AugChemception network with open source tools.
In addition to source code and package assembly instructions, Bjerrum's tutorial depicts examples of AugChemception images. For example, the following image illustrates variable bond shading and atom colorings:

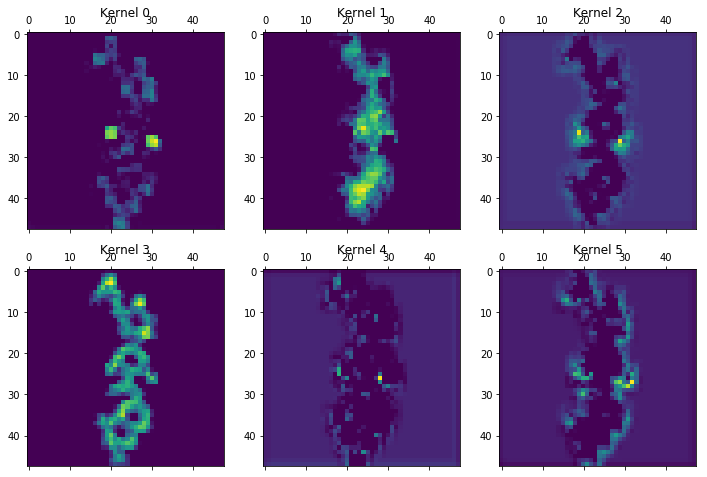
CNNs offer the capability to peek into various layers by examining the filters or "kernels" being used. The following example from layer 15 gives an idea of the features being detected by Chemception:
Conclusion
I doubt that the current accuracy of Chemception's predictions would be of practical use today. Rather, Chemception provides a platform from which such systems may eventually emerge. Recent history suggests that such an emergence may be closer than it seems.
Chemception offers a glimpse into a future in which lightly processed chemical datasets can be fed directly into off-the-shelf data learning pipelines to yield highly accurate predictive models. In this future, an iteratively hand-crafted molecular representation is no longer necessary. Instead, the system adapts itself to a much more raw form of structural data, identifying and classifying molecular features on its own and in a matter that may significantly diverge from human intuition.
Chemception also hints at the potential for deepening insights into numerous kinds of structure-property relationships. Already it's clear that some predictions will be more amenable to this streamlined approach than others. Could, for example, well-suited problems have some deeper, as yet unidentified, physical connection? Investigations along these lines may be aided by probing the convolutional layers generated during training.
Finally, it's worth mentioning the uncanny way in which Chemception seems to operate. For many chemists, and medicinal chemists in particular, 2D structure images are the bread-and-butter for the work they do. Structure images are information-rich, readily parsed, and instantly recognizable. They are the go-to resource for just about any project focused on getting small organic molecules to do something useful. The fact that Chemception is using a nearly identical form of input to perform what is essentially the same task as many highly-trained chemists is… unsettling.
It's one thing for a neural network to beat the pants off of someone classifying cat pictures or playing Go. It's an entirely different matter to ponder a neural network that routinely surpasses the world's best medicinal chemists.