Dealing with Delocalization
The valence bond (VB) model is a central abstraction in cheminformatics, underpinning everything from serialization formats to toolkit data structures. Like any approximation, the VB model makes certain simplifying assumptions. When one fails, trouble isn't far behind. This article describes the VB model's weakest assumption, and a conservative approach to avoiding the fallout from its near-certain invalidation.
The Delocalization Problem
The VB model's weakest assumption is localization. Under this assumption, a 1:1 association exists between an electron and a given atom pairing (aka "bond"), or alternatively between a proton and an atom. Exceptions involving both proton and electron localization can be found throughout organic chemistry.
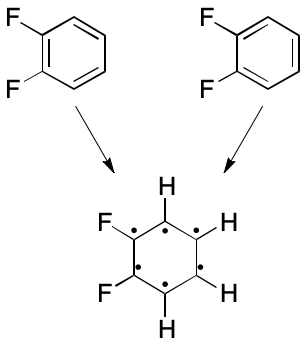
Consider 1,2-difluorobenzene. Two VB structures can be written. In one a double bond spans the two fluorene-bearing carbon atoms. In the other, the fluorene-bearing carbons are spanned by a single bond. The assumption in play is that each electron can be placed into one and only one two-atom bond. What happens instead is that electrons are distributed among all six ring atoms through an extended bonding arrangement. The VB model's blind spot toward multi-centered bonding leads to the observed distinction, which is an artifact.
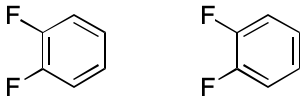
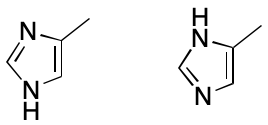
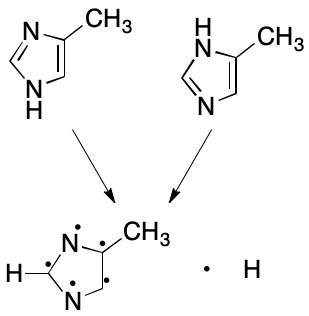
Next consider 4-methylimidazole. Once again, two VB structures can be written. In one a hydrogen atom is associated with the nitrogen proximal to the methyl-bearing carbon, and in the other the hydrogen atom is associated with the distal nitrogen atom. The assumption that fails here is that a proton can be associated with one and only one atom. What happens instead is that the hydrogen atom is associated with both nitrogen atoms. The VB model's failure to account for this kind of bonding leads to the two VB structures, whose differences are for most practical purposes artifactual. The problem is compounded in this example by the combined effects of proton and electron delocalization.
A previous article focused on this phenomenon from the perspective of electron delocalization only, referring to it as Delocalization-Induced Molecular Equality (DIME). As can be seen from the two examples above, however, delocalization applies to associations involving both electrons and protons. Although this phenomenon might go by different names, they are all just different faces of the same iceberg. Fortunately, this similarity can be exploited when developing a solution.
Delocalization's effects don't always need to be addressed, as further described in the next section. But when the breakdown of the VB model does become noticeable, the effects can be extremely disruptive. Molecular equality determinations are fertile ground for breeding such problems, with chemical registries being ground zero. However, many systems that don't look like registries on the surface are in fact registries.
The VB model fails because it assumes universal electron and proton localization, despite ample experimental evidence to the contrary. By adopting the VB model, cheminformatics serialization formats and toolkits pay the price for this bad assumption in the form of artifactual distinctions between equivalent encodings. What's needed is a way to deal with delocalization within a framework that uses localization.
Ignoring the Problem
Further insights can be gleaned from those contexts in which the delocalization problem can be ignored. Examples include molecular weight and formula determinations. Delocalization can be ignored here because bonding and connectivity are disregarded. In other words, both outputs can be computed from just a list of elements. Therefore, a sure way to eliminate localization artifacts is to disregard bonding.
Of course, this approach may only be partially feasible. Consider the computation of extended connectivity fingerprints. If implemented with care, electron delocalization artifacts can be avoided by replacing bond order with atomic subvalence. For an illustration, see this approach. Even so, proton delocalization will necessarily become a factor to the extent that the computation treats atomic hydrogen count as an invariant. And this includes a lot of computations.
Limited though they may be, these exceptional cases reveal something perhaps obvious but still important. Localization artifacts disappear when localization is impossible.
Solutions
Identifying the delocalization problem and some cases in which it can be ignored provides a foundation for evaluating solutions. These generally fall into one of two categories:
- Canonicalization. Designate one artifactually-equivalent form as privileged. This form need not adhere to experimental observations or even "chemical intuition." The only requirement is that the same form is generated from any equivalent encoding.
- Subsetting. Disable or remove those representation features that cause artifacts in the first place.
- Elevating delocalization. Add features to the encoding that explicitly take delocalization into account.
Canonicalization is the more limited approach. It can work for problems like exact structure match, but fails in any context that treats atomic hydrogen count, subvalence, or hybridization as an invariant.
A more general solution is subsetting. Abolishing the concept of localized protons and electrons eliminates artifactual effects at their source. The compatibility of canonicalization with the VB model is probably obvious. What may not be so clear is how subsetting can also be compatible.
The elevation of delocalization is a more unusual, but effective, approach. Perhaps the best-known platform to adopt it is InChI. Hydrogens can be explicitly associated with multiple atoms as "mobile hydrogens." A lesser-known option does something similar for electron delocalization through the introduction of the bonding system concept.
Subsetting the VB Model
A starting point for subsetting the VB model might include the following two changes:
- Allow bonds of order one only. Replace double, triple, and higher bond orders are replaced by single bonds. Electron delocalization occurs for the most part through multiple bonds. Eliminating multiple bonds makes this form of delocalization impossible. This step only works if atoms explicitly encode the hydrogens they are associated with.
- Transform all heteroatom-bound hydrogens to free hydrogens. A free hydrogen is a disconnected component consisting of an atomic hydrogen. Each hydrogen separated from its heteroatom yields one free hydrogen atom. Many cases of proton delocalization occur through heteroatom-heteroatom transfer. Eliminating hydrogen atoms from heteroatoms makes this form of delocalization impossible.
Consider how subsetting affects the representation of 1,2-difluorobenzene. All bonds have order one. The two fluorene-bearing carbons have zero associated hydrogens, and the rest have one associated hydrogen. Molecular formula and molecular mass can both be computed accurately, as can other descriptors. But now, only one representation is possible.
Subsetting also reduces the number of representations of 4-methylimidazole to one. Each nitrogen atom is associated with zero hydrogens. The molecule as a whole, however, is associated with one free hydrogen. These changes collapse the number of possible representations to one.
The main advantage of subsetting is that most tools designed to work prior to the change work afterword as well. This is by design. Adding no new features but rather just subtracting them means that file formats and internal representation continue to work without error under the new rules.
Variations
The subsetting procedure outlined here is too simple to be applied across the board. For example, it disregards the interplay between formal charge and protonation. Consider the 4-methylimidazole anion. Some contexts (e.g., a registry) require equivalence of deprotonated anion and protonated neutral parent forms. Others (e.g., electron density) consider each form distinct. Applications requiring equivalence between the anion and its parent can ensure this with a modification to the subsetting rules: for each negatively charged heteroatom, subtract a valence electron and add a free hydrogen. Application of this rule yields the same representation for both 4-methylimidazole and its deprotonated form.
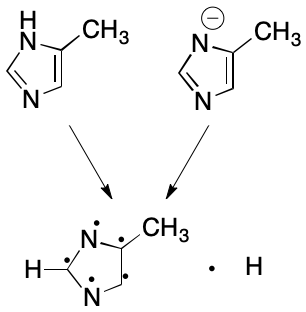
Likewise, the protonated form of 4-methylimidazole can be made equivalent to the parent with one more modification: for each positively charged heteroatom, remove a hydrogen, leaving its electron on the heteroatom. If no hydrogen is available, do nothing. Do not add a free hydrogen in either case.
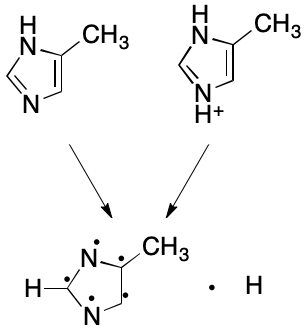
Tradeoffs
Subsetting can result in various side-effects, depending on the environment. For example, Balsa treats directional bonds (/ and \) not adjacent to doubles bonds as errors. Removal of the double bond entirely can therefore result in a downgrade of directional bonds, which eliminates the ability to differentiate double bond conformers. In contrast, molfiles are less influenced because conformation is set by atomic coordinates. But this is hardly universal. V3000 molfiles defined double bond conformation purely as a function of atomic coordinates, whereas V2000 molfiles support a double bond stereo attribute that can be set to "cis or trans." Similarly, conformational descriptors may be invalidated through the removal of an associated hydrogen atom. However, a case could be made that hydrogen-bearing heteroatoms are unsuitable as stereocenters to begin with.
As described here, subsetting does not address enol-like tautomerism. However, it does provide a foundation for doing so. Given the keto form, any enol form can be generated and then handled by subsetting to arrive at the same output structure regardless of input. The same cautions about stereocenter invalidation apply.
References
The protocol for subsetting described here is closely related Sayle Hashing. As defined by Noel O'Boyle:
I’ll call this method Sayle Hashing (after all, this fits with the nautical theme of the title). In this particular case, the Sayle Hash consists of two parts, a SMILES string and an integer. The integer is the total of the formal charges on the scaffold minus the number of hydrogens on each non-carbon atom, while the SMILES string is the canonical SMILES for the scaffold after setting all bond orders to 1 and hydrogen counts to 0. An example may be useful at this point. …
Noel later reported an implementation in Open Babel.
Roger Sayle is of course the author of the well-known So you think you understand tautomerism? from which some of this article's concepts were drawn.
Conclusion
As famously noted by George Box, "All Models are wrong, but some are useful." The VB model is wrong because its fundamental assumption, localization, breaks down so often. Nevertheless, the VB model remains as pervasive today as five decades ago. This article proposes a unified framework for reasoning about and resolving the VB model's most troublesome artifacts in cheminformatics: electron delocalization and prototropy (e.g., tautomerism). Central to this approach is "subsetting," the identification and removal those representation features yielding the artifacts in the first place. The tools presented here come with some important caveats and as such are best described as "a good starting" point rather than a finished framework for addressing the many limitations of the VB model.